RNA-seq analysis: our interview to Brian haas.
Posted on 08 March, 2017 by Marianna Pauletto
RNA-Seq (RNA sequencing), has revolutionized the manner in which eukaryotic transcriptomes are analysed, at an unprecedented scale and speed, even in non-model organisms. Several tools are now available for de novo assembly of RNA-Seq, and among them Trinity is surely one of the most used and cited.
We have had the possibility to discuss with Brian Haas about RNA-Seq and Trinity.
Brian is a Senior Computational Biologist at the Broad Institute - and author of Trinity a de-novo RNA-Seq transcript assembler- and is the instructor of our RNA-seq Workshop in June: https://www.physalia-courses.org/courses/course11/
1. In the early 2000’s, the microarray technology opened the doors to high-throughput gene expression analysis, now it has been almost completely replaced by RNA-seq. Can you briefly describe the reasons why RNA-sequencing overcame the microarray gene expression analysis?
BH: There are a number of reasons for this: improved dynamic range for measuring gene expression, single base resolution of expressed transcripts, allowing for identification of novel transcripts (missing as targets on arrays), and more versatile applications of rna-seq including transcript isoform reconstruction and polymorphism discovery.
2. Although RNA-seq is the gold standard for transcriptome sequencing and gene expression analysis, there are still a few bottlenecks. Which are the main ones?
BH: In the case of non-model organisms, the de novo transcriptome assembly can be a bottleneck, sometimes taking days of computation to complete on a high-memory high-performance server.
3. RNA-seq is being extensively used also in non-model species. Which are the main issues when doing RNA-seq analysis with non-model species?
BH: High polymorphism rates and polyploidy can pose a number of challenges for assembly and downstream analyses. Efforts to minimize the genetic diversity of a sample are important to consider, such as sampling a single individual as opposed to a population.
4. Which are the most important points to keep in mind in the design of an RNA-seq experiment?
BH: You'll want sufficient read lengths for effective de novo transcriptome assembly, at least 75 base paired-end reads, sufficient depth of sequencing, and plenty of biological replicates for each of your samples in order to assess the biological variation in expression as needed for downstream statistical analysis of differential expression. Also, make all attempts to avoid batch effects.
5. How much does the transcriptome assembly affect the RNA-seq data analysis?
BH: More completely assembled transcripts should provide more accurate quantitation and functional annotation.
6. Do you think there is a standard pipeline for RNA-seq data analysis or the optimal strategy should be evaluated for each dataset?
BH: I would hope that any of the routinely used analysis pipelines would effectively capture the most salient aspects of the study. It's always a good idea to explore multiple approaches, though, to ensure that your findings aren't specific to some peculiar characteristic of a given system, and if they are, the results should be heavily scrutinized.
7. Which are the main benefits from using a suite analysis like Trinity?
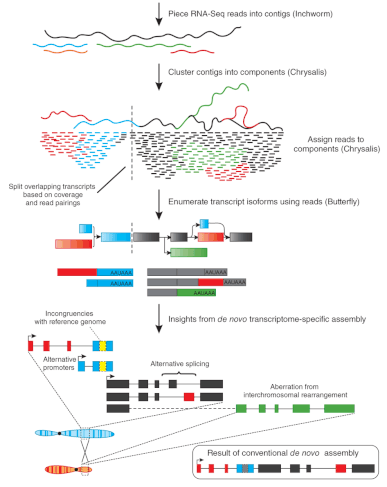
BH: Trinity was one of the first tools developed to tackle de novo
transcriptome assembly from RNA-Seq data. Now, there are several highly effective tools available. A benefit of using Trinity is that it includes
support for a variety of downstream analyses with plugins to other popular methods for transcript quantitation, differential expression, and functional annotation and analysis. Trinity has a
large global community of users and is actively supported by the community. As open source software, Trinity regularly has development contributed to
it from the greater bioinformatics community of developers. The Trinity documentation is extensive, with access to tutorials and
publications. Also Trinity assembly can be easily accessed on freely available compute resources. All
Trinity-related resources can be accessed via the Trinity website: http://trinityrnaseq.github.io